A novel platform to study the role of histone residues in animals
Post-translational modification of histone residues is thought to be the central mechanism of epigenetic regulation for many DNA-dependent processes. However, the role of histone post-translational modifications (PTMs) in animal cells has been inferred indirectly from mutations in the enzymes that catalyze a PTM (also known as “writers”) and the proteins that bind to and mediate the effect of a PTM (“readers”). We have established a genetic platform in Drosophila for generating and analyzing any desired histone genotype by using BAC-based arrays of histone genes to complement deletion of the endogenous histone locus (Figure 1). Due to the genomic organization of its replication-dependent histone genes and the sophisticated suite of tools available for genetic analysis, Drosophila is the only model organism in which a gene replacement strategy can be used to directly test the role of histone genes in animals. This work is part of a broader collaborative project with the Duronio, Matera, and Strahl labs at UNC (see Links).
Questions we ask:
Which histone residues are required for transcriptional regulation? Is the histone code required for developmental gene regulation?
Do histone PTM writer mutant phenotypes match those of mutant histone residues? If not, why?
Do the replication-independent histones perform similar functions as the replication-dependent histones?
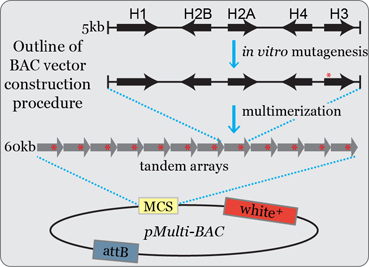
Figure 1. Multimerization of histone gene arrays for BAC-dependent transgenesis.
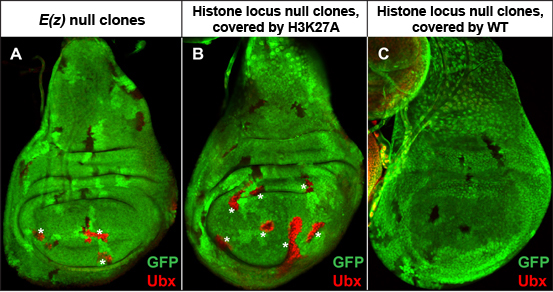
Figure 2. H3K27 is required for Polycomb target gene repression. (A) Mitotic clones of Enhancer of Zeste (E(z)), the writer of H3K27me3, show de-repression of the Polycomb target gene Ubx (red) in wing imaginal discs. (B) Histone locus mutant clones covered by H3K27A transgenes also de-repress Ubx. (C) Histone locus mutant clones covered by wild-type histone transgenes do not de-repress Ubx. Mutant clones are indicated by loss of GFP signal.
Recent Relevant Publications:
Leatham-Jensen M, Uyehara CM, Strahl BD, Matera AG, Duronio RJ, McKay DJ. Lysine 27 of replication-independent histone H3.3 is required for Polycomb target gene silencing but not for gene activation. PLoS Genetics. 2019 Jan;15(1):e1007932. PMCID: PMC6370247.
Armstrong RL, Penke TJR, Strahl BD, Matera AG, McKay DJ, MacAlpine DM, Duronio RJ. Chromatin conformation and transcriptional activity are permissive regulators of DNA replication initiation in Drosophila. Genome Research. 2018; 28(11). PMID: 30279224
Penke TJR, McKay DJ, Strahl BD, Matera AG, Duronio RJ. Functional Redundancy of Variant and Canonical Histone H3 Lysine 9 Modification in Drosophila. Genetics. 2018; 208(1):229-244. PMCID: PMC5753860
Meers MP, Henriques T, Lavender CA, McKay DJ, Strahl BD, Duronio RJ, Adelman K, Matera AG. Histone gene replacement reveals a post-transcriptional role for H3K36 in maintaining metazoan transcriptome fidelity. Elife. 2017 Mar 27;6. pii: e23249. doi: 10.7554/eLife.23249. PMCID: PMC5404926.
Penke TJ, McKay DJ, Strahl BD, Matera AG, Duronio RJ. Direct interrogation of the role of H3K9 in metazoan heterochromatin function. Genes & Development. 2016; 30(16):1866-80. PMID: 27566777.
McKay DJ, Klusza S, Penke TJR, Meers MP, Curry KP, McDaniel SL, Malek PY, Cooper SW, Tatomer DC, Lieb JD, Strahl BD, Duronio RJ, Matera AG. Interrogating the function of metazoan histones using engineered gene clusters. Developmental Cell. 2015; 32: 373-386. PMID 25669886.
